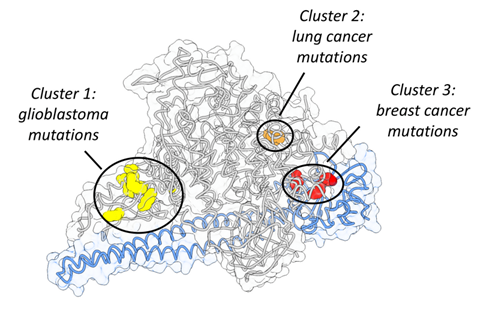
Three cancer mutation groups in the protein PIK3CA identified by different subgene algorhytms.
Researchers from the Barcelona Supercomputing Center are teaming up to evaluate the tools used to probe the cancer genome.
Eduard Porta-Pardo, a senior researcher in the Life Sciences Department at BSC has undertaken the first ever comparative analysis of sub-gene algorithms that mine the genetic information in cancer databases. These powerful data-sifting tools are helping untangle the complexity of cancer, and find previously unidentified mutations that are important in creating cancer cells.
“Finding new cancer driver genes is an important goal of cancer genome analysis,” adds Porta-Pardo. This study should help researchers understand the advantages and drawbacks of sub-gene algorithms used to find new potential drug targets for cancer treatment.
The study, published today in Nature Methods, reviews, classifies and describes the strengths and weaknesses of more than 20 algorithms developed by independent research groups. The evaluation of cancer genome analysis methods is a key activity for the selection of the most adequate strategies to be integrated in the BSC’s personalized medicine platform.
Despite the increasing availability of genome sequences, a common assumption is to consider a gene as a single unit, however, there are a number of events, such as DNA substitutions, duplications and losses that can occur within a gene—at the sub-gene level. Sub-gene algorithms provide a high-resolution view that can explain why different mutations in the same gene can lead to distinct phenotypes, depending on how the mutation impacts specific protein regions. A good example of how different sub-gene mutations influence cancer is the NOTCH1 gene. Mutations in certain regions of NOTCH1 cause it to act as a tumor suppressor in lung, skin and head and neck cancers. But, mutations in a different region can promote chronic lymphocytic leukemia and T cell acute lymphoblastic leukemia. So it is incorrect to assume that mutations in a gene will have the same consequences regardless of their location.
The study researchers applied each sub-gene algorithm to the data from The Cancer Genome Atlas (TCGA), a large-scale data set that includes genome data from 33 different tumor types from more than 11,000 patients. “Our goal was not to determine which algorithm works better than another, because that would depend on the question being asked,” says Eduard Porta-Pardo, first author of the paper. “Instead, we want to inform potential users about how the different hypotheses behind each sub-gene algorithm influences the results, and how the results differ from methods that work at the whole gene level.” Porta-Pardo is a former postdoc at Sanford Burnham Prebys Medical Discovery Institute (SBP) who has recently joined Life Sciences Department at BSC under the Director of Alfonso Valencia, also coauthor of this work.
The researchers have made two important discoveries. First, they found that the algorithms are able to reproduce the list of known cancer genes established by cancer researchers—validating the sub-gene approach and the link between these genes and cancer. Second, they found a number of new cancer driver genes—genes that are implicated in the process of oncogenesis—that were missed by whole-gene approaches.
Although Sanford Burnham Prebys Medical Discovery Institute (SBP) has led the project, this paper has been the effort between international institutions as Harvard Medical School, Institute for Research in Biomedicine (IRB), Universitat Pompeu Fabra (UPF) & Spanish National Cancer Research Centre, among others.

