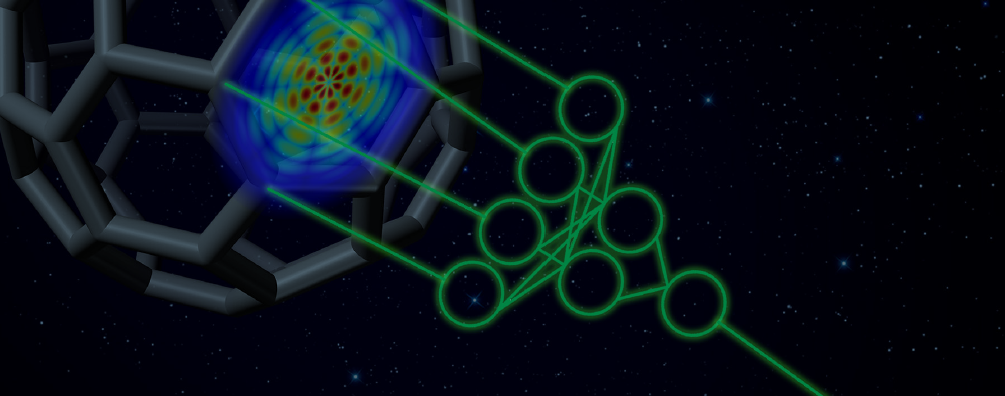
This sponsored post from NVIDIA is the third of five in a series of case studies that illustrate how AI is driving innovation across businesses of every size and scale. This article explores how the University of Florida and University of North Carolina developed an anakin-me neural network engine to produce computationally fast quantum mechanical simulations with high accuracy at a very low cost.
In the pharmaceutical industry, drug discovery is a long and expensive process. It takes an average of 12 years and $2.6 billion to bring a new drug to market. One key to speeding the drug discovery process is the ability to more accurately simulate molecular dynamics (MD), to quickly screen millions of potential drug combinations so researchers can focus their energy on the most promising options.

ANI-1 is within 1 kcal/mol from reference DFT calculations on much larger molecular systems than those included in the training data set.
All drug discoveries require molecular simulations to understand their potential efficacy. Molecular energetics, where millions of molecules are scanned to determine how they interact with each other, helps in this understanding. However, to have accurate MD simulations, you need accurate quantum mechanical (QM) simulations as well. QM simulations are essential to the process of accurately screening millions of potential drugs.
Unfortunately, QM simulation is so computationally expensive that researchers are pressured to use less accurate approximations, resulting in less effective drug candidates. Accurate molecular energy calculations using DFT scale with the cube power (O(N3)) of the system size. Therefore, accurate calculations are restricted to relatively small systems and short time scales for MD simulations. Adding to it all, screening 10 million drug candidates with QM simulation can take up to five years to compute. It was this slow, expensive process that brought together researchers at the University of Florida (UFL) and the University of North Carolina (UNC), who were determined to increase the accuracy of QM simulations while at the same time reducing the process from years to mere minutes.
ANI is a deep neural network that learns QM energy function to produce computationally fast and very accurate molecular energy surfaces, geometries, and forces.
Deep Learning for QC Simulation
To solve the problem of QM simulation time and cost, Justin S. Smith, a chemistry graduate student at UF working under the supervision of Dr. Adrian Roitberg, UF Department of Chemistry, and Dr. Olexandr Isayev, UNC Eshelman School of Pharmacy, developed a new simulation procedure and method called ANAKIN-ME (Accurate NeurAI network engINe for Molecular Energies) or ANI for short. ANI is a deep neural network that learns QM energy function to produce computationally fast and very accurate molecular energy surfaces, geometries, and forces.
Today the total number of calculated molecular structures is over 20 million. ANI uses deep learning with NVIDIA GPUs to predict molecular energy surfaces for molecules as accurate as methods that are six orders of magnitude more computationally expensive. ANI is also transferable to any organic molecule with C, H, N, O, S, F atoms, which covers a good portion of the chemistry/medicinal, chemistry/drug discovery space, and more elements are on the way. Also, ANI can break bonds and describe chemical reactions, overcoming one of the biggest limitation of modern force fields.
Essentially, ANI is trained to learn Hamiltonian of the Schrodinger equation, and the results are almost instantaneous. ANI’s accuracy is approximately ~1 kcal/mole from reference DFT data which is within chemical accuracy. ANI can also reproduce the first-principles model for QC simulations, delivering equivalent chemical accuracy at 1,000,000x the speed. Finally, ANI can screen 10 million drug candidates in just 8 minutes with NVIDIA GPUs, versus 240 days on CPU.
“A fast and accurate description of molecular energetics can lead to breakthroughs in many fields, including drug discovery and materials science. ANI may start the next revolution in computational chemistry.” – Justin Smith, Graduate Student, Chemistry, University of Florida
Faster Time to Market
ANI opens up a whole new world for drug discovery and materials science. Leveraging ANI, very large systems, and time scales will become routinely accessible through deep learning simulations. More importantly, faster and more accurate screening of new drugs is now possible, and costs are significantly reduced by eliminating the expense of synthesizing molecules and running numerous experiments.
“A fast and accurate description of molecular energetics can lead to breakthroughs in many fields including drug discovery and materials science. ANI may start the next revolution in computational chemistry,” Justin S. Smith a Chemistry Graduate student at the University of Florida.
[clickToTweet tweet=”With ANI, discovery possibilities are endless. #AI #quantum ” quote=”With ANI, discovery possibilities are endless. #AI #quantum “]
With ANI, discovery possibilities are endless. ANI prevails where cheaper methods fail and where true MD is needed but is cost prohibitive. Whether it is modeling solar cells for higher efficiency or solving what is called the polymorph problem in late drug discovery, ANI can predict changes in material properties, give greater understanding of molecular interactions and in the end help bring products to market faster than ever before.
Additional case studies in this series cover the following topics:
- DeepSat: Monitoring the Earths Vitals with AI
- GPUs Accelerate Population Distribution Mapping Around the Globe
This case study on innovation within the AI industry from NVIDIA first ran as part of the company’s Deep Learning Success Stories.

