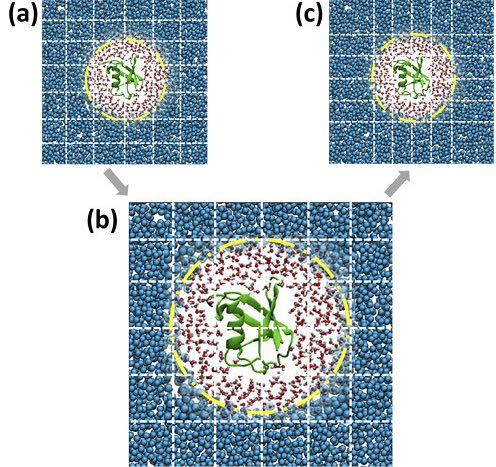
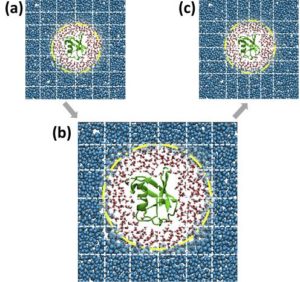
Adaptive resolution situation (AdResS) simulation of an atomistic protein, its atomistic hydration shells and CG water particles. The dashed lines illustrate the spatial domain decomposition. When going from (a) to (c), the effects for load imbalance get mitigated.
Researchers at Los Alamos National Lab have developed new software to distribute computation more efficiently and across increasing numbers of supercomputer processors. This new decomposition approach for molecular dynamics simulation is called the heterogeneous spatial domain decomposition algorithm, or HeSpaDDA. Areas of different density were assessed and rearranged to distribute the processing workload.
In this article, we introduce the Heterogeneous Spatial Domain Decomposition Algorithm (HeSpaDDA). This algorithm is not based in any runtime measurements, it is rather a predictive computational resources allocation scheme. The HeSpaDDA algorithm is a combination of an heterogeneity (resolution or spatial density) sensitive processor allocation with an initial rearrangement of subdomain-walls. The initial rearrangement of subdomain-walls within HeSpaDDA anticipates favorable cells distribution along the processors per simulation box axis by moving cell boundaries according to the resolution of the tackled region in the molecular system. In a nutshell, the proposed algorithm will make use of a priori knowledge of the system setup. Specifically, the region which is computationally less expensive. This inherent load-imbalance could come from different resolutions or different densities. The algorithm will then propose nonuniform domain layout, i.e. domains of different size and its distribution amongst compute instances. This could lead to significant speedups for systems of the aforementioned type over standard algorithms.
LLNL is involved in several supercomputing efforts, including the Exascale Computing Project and machine procurement for the Advanced Simulation and Computing program. The key challenge to these efforts is effectively utilizing these machines at scale. For instance, as part of ECP, there are ongoing efforts that range from designing the hardware to over-building the ECP software components to writing the next application codes. On the application side, one key component is to ensure that the computational load is well-balanced between all computing units.
The method was tested for two different molecular dynamics setups with inherent load-imbalance. In one case, the protein ubiquitin’s behavior in water in an adaptive resolution situation (AdResS) was examined. A speedup of a factor 1.5 was measure for that system (see figure for illustration). The second system studied was a model fluid system with two phases (a Lennard-Jones binary fluid) and a speed up of a factor 1.23 was measured.
This work began under a LDRD postdoctoral Director’s fellowship and was finished as part of the exascale-computing program (ECP) Co-Design Center for Particle Applications (CoPA) led by Tim Germann of the Lab’s Physics and Chemical Material’s group.
The work was done in collaboration with researchers, led by Horacio Vargas Guzman, from the Max Planck Institute for Polymer Research (MPI-P) in Mainz, Germany. At the same institute, AdResS was pioneered by Kurt Kremer’s group, which is a method that divides simulations into areas of high- and low-resolution, based on how much information and complexity is needed in each area.
The work was featured in both the Physical Review E journal and part of the May 2018 issue of ASCR Discovery.

